業務說明
- 更新日期:2024-05-23
- 點閱次數:938
藥品技術性資料審查
財團法人醫藥品查驗中心(以下簡稱查驗中心)接受衛生褔利部食品藥物管理署委託,協助進行藥品上市前與上市後相關的技術性資料評估。
一、藥品上市前審查重點
藥品上市前的審查,包含了臨床試驗階段的試驗計畫審查、藥品生體可用率試驗計畫書及報告審查、銜接性試驗評估與申請上市的藥品查驗登記。其中藥品查驗登記又可分為新藥查驗登記審查、學名藥查驗登記審查、符合指示藥品審查基準之藥品查驗登記審查及原料藥查驗登記審查。為確保民眾用藥安全,本中心針對具安全性考量(如有重大已知風險或潛在風險)的藥品進行風險管理計畫及執行成效報告審查。此外,為提升國內製劑使用的原料藥品質管理,查驗中心亦協助衛生福利部食品藥物管理署進行原料藥主檔案之技術性資料評估。
(一)臨床試驗計畫審查時,主要著重於確保受試者參與試驗設計良好的臨床試驗,使用具有品質管控的藥品,試驗的過程中,能有足夠的安全性監測與保護措施,以保障受試者的權益。
(二)藥品生體可用率/生體相等性試驗計畫書及報告書審查需評估:
-
- 執行該生體相等性試驗之目的與對照品選擇之合適性
- 比較試驗藥品與對照藥品之化學、製造與管制基本資料
- 試驗設計合理性
- 統計分析方法
試驗報告書另包含
-
- 檢品含量分析方法確效
- 試驗檢品分析
- 試驗統計結果
(三)銜接性試驗評估乃藉由藥品之藥動/藥效學或療效、安全資訊,輔以比較東亞族群(例如台灣)與非東亞族群的數據,評估國外臨床試驗數據是否能外推至我國,以利評估我國病人之用法用量的合理性。
(四)新藥查驗登記的技術性資料評估重點著重於:
-
- 化學、製造與管制的資料能顯示藥品的原料藥與製劑品質有良好的控管,於不同批次之間,具有穩定的品質一致性。
- 動物的藥理與毒理資料能支持藥品的作用機制,可完整評估可能的潛在毒性反應。
- 由動物與人體的藥動/藥效學資料可了解藥品基本藥動/藥效學特性,且由藥品於特殊族群的藥動學資訊,與其他藥品的交互作用資訊,有利於評估藥品的於特殊族群及與其他藥品合併使用時,用法用量調整的合理性。
- 臨床試驗的結果能顯示藥品於宣稱適應症的族群具有可信的療效、與可接受的安全性,以支持宣稱用法用量的合理性。
(五)學名藥查驗登記的技術性資料評估重點著重於:
-
- 化學、製造與管制的資料能顯示學名藥藥品與對照藥品的品質相當,其原料藥與製劑品質有良好控管,於不同批次之間,具有穩定的品質一致性。
- 藥動技術性資料,例如:生體相等性試驗的資料,能顯示學名藥藥品與對照藥品間具有生體相等性,以支持學名藥品與對照藥品有相當的療效及安全性;或在符合一定前提下,得以體外比對試驗資料(溶離率曲線比對試驗或其他試驗方式),作為支持學名藥品與對照藥品具有生體相等性。
(六)符合指示藥品審查基準的藥品,因其所含主成分之療效與安全性已獲確認,因此主要著重於化學、製造與管制的資料能顯示藥品的品質有良好的控管,且於不同批次之間,具有穩定的品質一致性。
(七)風險管理計畫審查重點,著重於是否已充分向病人及醫療人員說明藥品可能風險、風險預防及降低措施的適當性、風險管理計畫執行方式及評估成效方式;風險管理計畫執行成效報告審查重點,則著重於執行成效評估、風險管理計畫遵從性評估、藥品安全監視成效評估。
(八)原料藥查驗登記/原料藥主檔案之技術性資料評估,著重於原料藥在鑑別、含量、純度及與安全性相關之品質特性,以支持藥品之品質、安全與療效。
二、上市後藥品變更登記
藥品上市之後,若登記事項變更,應依規定申請變更許可。查驗中心協助食品藥物管理署進行之已上市藥品申請變更案件中,涉及仿單、適應症、用法用量、藥品類別、製造場所、劑型、檢驗規格與方法、賦形劑、直接包裝材質等變更之技術性資料評估。
因應不同的變更類別,審查的重點亦不同。以適應症、用法用量之變更而言,通常著重於臨床試驗資料是否足以支持所宣稱之變更。若為製程、製造場所或賦形劑變更,則著重於變更後與變更前藥品的品質是否維持不變,變更前後的藥品是否具生體相等性或體外溶離率曲線是否相似。若檢驗規格與方法變更,則主要考量變更的依據是否合理,相關改變是否仍可以確保藥品的品質控管。符合指示藥品基準之藥品的上市後變更,主要針對適應症、用法用量、包裝種類、類別、仿單內容進行審查。
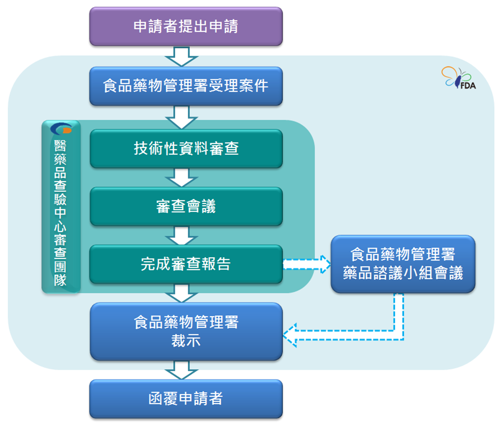
三、作業流程說明
申請者於衛生福利部食品藥物管理署藥品組收發窗口送件後,由行政審查員負責行政作業及專案管理,並啟動組成審查團隊;團隊成員依案件類別可包括:化學製造管制、藥毒理、藥動藥效、統計與臨床等專業。行政審查員視需要,將根據標準作業流程召開審查團隊會議,由Team Leader主持,就案件之技術性資料進行討論,以完成審查報告及結論。審查團隊完成審查報告後,必要時得提至衛生福利部食品藥物管理署藥品諮議小組/再生醫學諮議小組/罕見疾病及藥物審議委員會討論。最後由行政審查員彙整審查報告及委員會會議結論,進行行政呈核與裁示。